# FastClone
FastClone is a fast algorithm to infer tumour heterogeneity. Given somatic
mutation frequencies and copy number data, FastClone infers subclonal
composition and phylogeny. The algorithm won the first place in DREAM Somatic
Mutation Calling -- Heterogeneity Challenge.
# Installation
FastClone needs Python 3.5 or later version. It needs logbook, python-fire,
scikit-learn, and pandas. To install the package using Pip,
```
git clone https://github.com/GuanLab/FastClone_GuanLab.git
pip install FastClone_GuanLab/
```
(Please make sure you have the slash at the end, which forces pip to install from local directory, otherwise it will run into error)
You also can directly pip install FastClone with the command below.
```
pip install fastclone-guanlab
```
# Usage
FastClone accepts either MuTect VCF + Battenberg format (specified in the DREAM
SMC-Het Challenge) or PyClone format.
The general format of command line for reading PyClone format files:
```
fastclone load-pyclone prop [FILE_NAME] [TUMOR_PURITY] solve [OUTPUT_PATHWAY]
```
(If purity is unavailable, input "None" at the position of [TUMOUR__PURITY], and FastClone will infer purity automatically)
A pseudo example to load samples from PyClone format files and infer (t1.tsv is included in this repository):
```
fastclone load-pyclone prop t1.tsv 0.8 solve ./fastclone_result
```
(Please make sure t1.tsv is under your current directory. Note this pseudo example only has one clone with a purity ~0.15)
The general format of command line for reading VCF + Battenberg format files:
```
fastclone load-mutect-battenberg prop [VCF_FILE_NAME] [TUMOR_/_NORMAL_COLUMN] [BATTENBERG_FILE_NAME] solve [OUTPUT_PATHWAY]
```
A pseudo example to load samples from VCF + Battenberg files (VCF + Battenberg mode does not need tumor purity):
```
fastclone load-mutect-battenberg prop 0009b464-b376-4fbc-8a56-da538269a02f tumor 0009b464-b376-4fbc-8a56-da538269a02f.consensus.20170119.somatic.cna.txt solve ./fastclone_result
```
(Example files are in the folder called Battenberg_VCF_sample)
Run `fastclone` for more help information.
If MuTect VCF and PyClone samples are provided, note that MuTect
mutations are labelled as 'Chromosome:Coordinate:AltBase', such as
'Y:15989697:G'. Make sure PyClone ID uses the same ID.
Separately, subclone.py will infer purity (whether a starter value is given or not), and subclone identification and assignment; phylogeny.py will infer phylogeny.
# Output
1.subclones.csv gives proportion of each clone in a tumor sample.
2.scores.csv gives SNPs assignment. Each column corresponding to a clone, and the entries in each column indicates how likely the SNP is assigned to the clone.
3.phylogeny.png shows the tree structure of clones.
4.phylogeny_proportion.csv gives the proportion of a subclone in its parent clone.
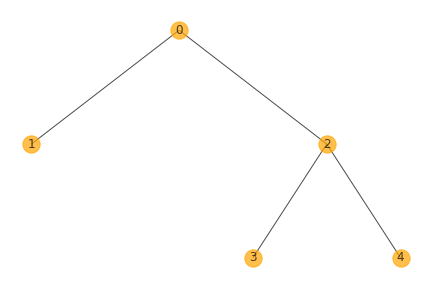
(We named clones with numeric values, which starts from 0, and the names are consistent within all output files)
# Versions
## 1.0.9
* Fix the phlogeny module, and add a new function to output the proportion of a subclone in its parent clone.




