<p align="center"><img src="https://raw.githubusercontent.com/TurtleTools/caretta/master/caretta_logo.png" width="300" title="Caretta Logo"></p>
[](https://badge.fury.io/py/caretta)
[](http://dx.doi.org/10.1016/j.csbj.2020.03.011)
[](http://dx.doi.org/10.1101/2021.04.07.438777)
# Caretta-shape – A multiple protein structure alignment and feature extraction suite
Caretta is a software-suite to perform multiple protein structure alignment and structure feature extraction.
Visit the [demo server](http://bioinformatics.nl/caretta) to see caretta's capabilities. The server only allows alignment of up to 50 proteins at once. (This is currently down, will be back up soon!)
The command-line tool and self-hosted web application do not have this restriction.
The older, slower version of Caretta as described in https://doi.org/10.1016/j.csbj.2020.03.011 can be found at https://git.wur.nl/durai001/caretta
## Installation
### Requirements
#### Operating system support
1. Linux and Mac
* All capabilities are supported
2. Windows
* The external tool **msms** is not available in Windows. Due to this:
* Feature extraction is not available.
* `features` argument in caretta-cli must always be run with `--only-dssp`.
* `caretta-app` is not available.
#### Software
Caretta works with Python 3.7+
Run the following commands to install required external dependencies (Mac and Linux only):
```bash
conda install -c salilab dssp
conda install -c bioconda msms
```
### Install both the command-line interface and the web-application (Mac and Linux only):
```bash
pip install "caretta[GUI] @ git+https://github.com/TurtleTools/caretta.git"
```
### Install only the command-line interface:
```bash
pip install git+https://github.com/TurtleTools/caretta.git
```
### Environment variables:
```bash
export OMP_NUM_THREADS=1 # this should always be 1
export NUMBA_NUM_THREADS=20 # change to required number of threads
```
## Usage
### Command-line Usage
```bash
caretta-cli input_pdb_folder
# e.g. caretta-cli test_data
```
Options:
```
Usage: caretta-cli [OPTIONS] INPUT_PDB
Align protein structures using Caretta.
Writes the resulting sequence alignment and superposed PDB files to
"caretta_results". Optionally also outputs a set of aligned feature
matrices, or the python class with intermediate structures made during
progressive alignment.
Arguments:
INPUT_PDB A folder with input protein files [required]
Options:
-p FLOAT gap open penalty [default: 1.0]
-e FLOAT gap extend penalty [default: 0.01]
-c, --consensus-weight FLOAT weight well-aligned segments to reduce gaps
in these areas [default: 1.0]
-f, --full Use all vs. all pairwise alignment for
distance matrix calculation (much slower)
[default: False]
-o, --output PATH folder to store output files [default:
caretta_results]
--fasta / --no-fasta write alignment in FASTA file format
[default: True]
--pdb / --no-pdb write PDB files superposed according to
alignment [default: True]
-t, --threads INTEGER number of threads to use for feature
extraction [default: 4]
--features extract and write aligned features as a
dictionary of NumPy arrays into a pickle
file [default: False]
--only-dssp extract only DSSP features [default: False]
--class write StructureMultiple class with
intermediate structures and tree to pickle
file [default: False]
--matrix write distance matrix to file [default:
False]
-v, --verbose Control verbosity [default: True]
--install-completion [bash|zsh|fish|powershell|pwsh]
Install completion for the specified shell.
--show-completion [bash|zsh|fish|powershell|pwsh]
Show completion for the specified shell, to
copy it or customize the installation.
--help Show this message and exit.
```
### Web-application Usage (Mac and Linux only)
```bash
caretta-app <host-ip> <port>
# e.g. caretta-app localhost 8091
```
Then go to localhost:8091/caretta in a browser window.
### Features
* `dssp_NH_O_1_index`, `dssp_NH_O_1_energy`, `dssp_NH_O_2_index`, `dssp_NH_O_2_energy`, `dssp_O_NH_1_index`,
`dssp_O_NH_1_energy`, `dssp_O_NH_2_index`, `dssp_O_NH_2_energy`: hydrogen bonds; e.g. -3,-1.4 means: if this residue is residue i then N-H of I is h-bonded to C=O of I-3 with an
electrostatic H-bond energy of -1.4 kcal/mol. There are two columns for each type of H-bond, to allow for bifurcated H-bonds.
* `dssp_acc`: number of water molecules in contact with this residue *10. or residue water exposed surface in Angstrom^2.
* `dssp_alpha`: virtual torsion angle (dihedral angle) defined by the four Cα atoms of residues I-1,I,I+1,I+2. Used to define chirality.
* `dssp_kappa`: virtual bond angle (bend angle) defined by the three Cα atoms of residues I-2,I,I+2. Used to define bend (structure code ‘S’).
* `dssp_phi`: IUPAC peptide backbone torsion angles.
* `dssp_psi`: IUPAC peptide backbone torsion angles.
* `dssp_tco`: cosine of angle between C=O of residue I and C=O of residue I-1. For α-helices, TCO is near +1, for β-sheets TCO is near -1.
* `anm_ca`: Fluctuations of alpha carbon atoms based on an Anisotropic network model
* `anm_cb`: Fluctuations of beta carbon atoms based on an Anisotropic network model
* `gnm_ca`: Fluctuations of alpha carbon atoms based on a Gaussian network model
* `gnm_cb`: Fluctuations of beta carbon atoms based on a Gaussian network model
* `depth_ca`: Depths of alpha carbon atoms
* `depth_cb`: Depths of beta carbon atoms
* `depth_mean`: Mean depth of residues
## Publications
Janani Durairaj, Mehmet Akdel, Dick de Ridder, Aalt DJ can Dijk. "Fast and adaptive protein structure representations for machine learning." [Machine Learning for Structural Biology Workshop](mlsb.io), NeurIPS 2020 (https://doi.org/10.1101/2021.04.07.438777)
Poster:
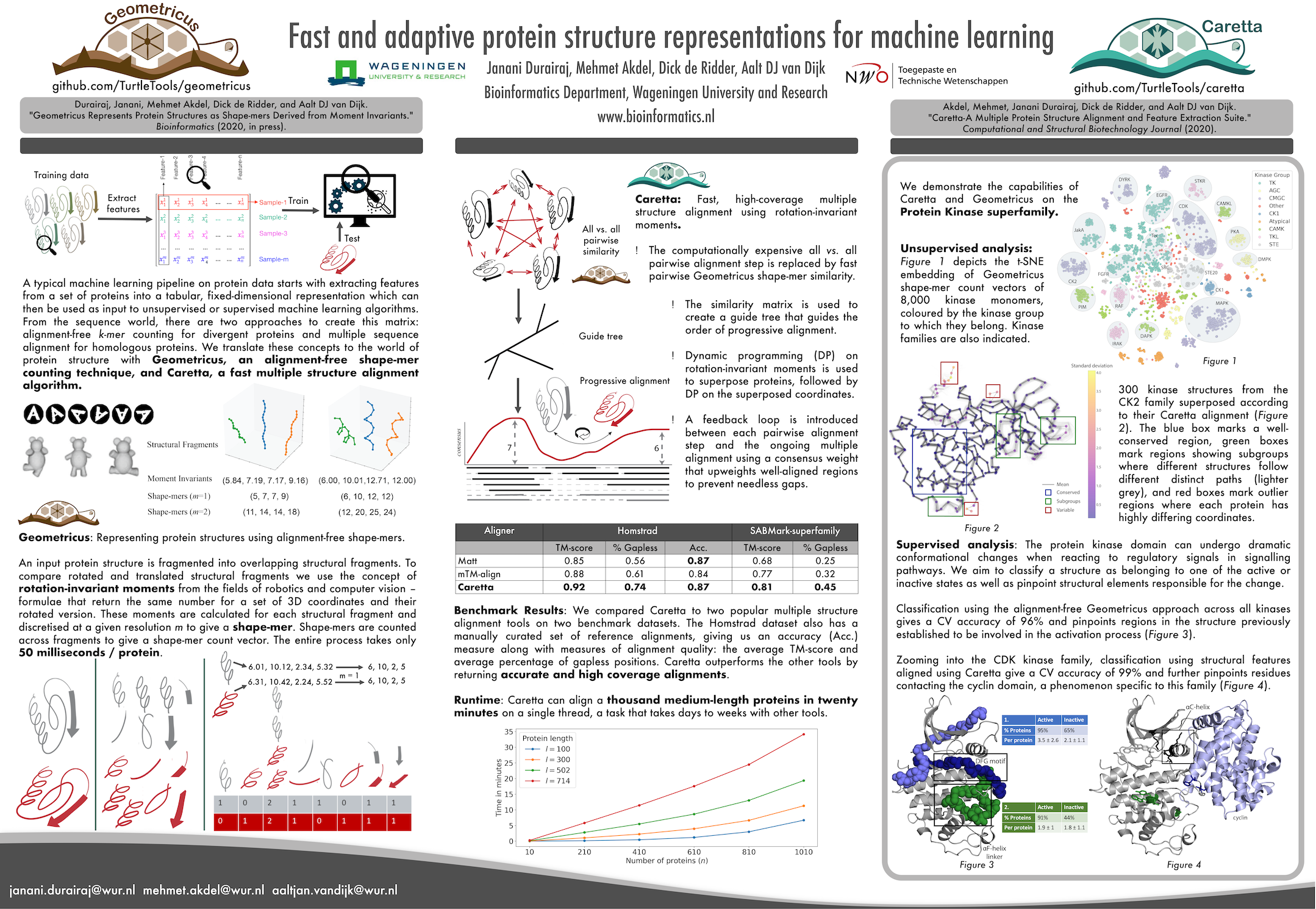
Akdel, Mehmet, Janani Durairaj, Dick de Ridder, and Aalt DJ van Dijk. "Caretta-A Multiple Protein Structure Alignment and Feature Extraction Suite." Computational and Structural Biotechnology Journal (2020). (https://doi.org/10.1016/j.csbj.2020.03.011)




